Epilepsy and Traumatic Brain Injury
The Biochemistry and Epigenetics of Epilepsy
Epilepsy, one of the most prevalent neurological conditions, presents as a complex disorder of network dysfunction characterized by spontaneous non-provoked seizures and associated comorbidities. Currently used antiepileptic drugs have been designed to suppress neuronal hyperexcitability and thereby to suppress epileptic seizures. However, the current armamentarium of antiepileptic drugs is not effective in over 30% of patients, does not affect the comorbidities of epilepsy, and does not prevent the development and progression of epilepsy (epileptogenesis). Prevention of epilepsy and its progression remains the Holy Grail for epilepsy research and therapy development, requiring novel conceptual advances to find a solution to this urgent medical need. The methylation hypothesis of epileptogenesis suggests that changes in DNA methylation are implicated in the progression of the disease. In particular, global DNA hypermethylation appears to be associated with chronic epilepsy. Clinical as well as experimental evidence demonstrates that epilepsy and its progression can be prevented by biochemical manipulations, and those that target previously unrecognized epigenetic functions contributing to epilepsy development and maintenance of the epileptic state.
Related research articles by Dr. Detlev Boison:
- The Biochemistry and Epigenetics of Epilepsy: Focus on Adenosine and Glycine
- Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis
- Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice

Focal Adenosine Augmentation Therapies to Treat Epilepsy
Research from our lab has demonstrated that deficiencies in the brain’s own adenosine-based seizure control system contribute to seizure generation. Consequently, the reconstitution of adenosinergic neuromodulation constitutes a rational approach for seizure control. Therefore, focal adenosine augmentation therapies (AATs) have significant potential for antiepileptic and disease-modifying therapy. Due to systemic side effects of adenosine focal adenosine augmentation – ideally targeted to an epileptic focus – becomes a therapeutic necessity. This has experimentally been achieved in kindled seizure models as well as in post status epilepticus models of spontaneous recurrent seizures using four different therapeutic strategies: (i) Polymer-based brain implants that were loaded with adenosine; (ii) Brain implants comprised of cells engineered to release adenosine and embedded in a cell-encapsulation device; (iii) Direct transplantation of stem cells engineered to release adenosine; and (iv)Knockdown of ADK in vivo using viral gene therapy vectors. To meet the therapeutic goal of focal adenosine augmentation, genetic disruption of the adenosine metabolizing enzyme adenosine kinase (ADK) in rodent and human cells in vitro (ex vivo gene therapy) or directly in vivo (in vivo gene therapy) was used as a molecular strategy to induce focal adenosine augmentation, which demonstrated potent antiepileptic and neuroprotective properties.
To read Dr. Detlev Boison’s research, click here.
Therapies for Epilepsy Prevention
Epilepsy and its progression can be prevented by the transient augmentation of adenosine. We have shown that overexpression of ADK and resulting adenosine deficiency and epigenetic changes play a major causal role in epileptogenesis. Our past research efforts have demonstrated that both focal adenosine augmentation through local adenosine-releasing silk- or cell-based brain implants, as well as systemic adenosine augmentation through a small molecule ADK inhibitor can prevent epilepsy or its progression in etiologically different rodent systems that model epileptogenesis in temporal lobe epilepsy (TLE), our clinical target. We have shown that therapeutic adenosine augmentation prevents kindling epileptogenesis in the rat, prevents epilepsy development in the mouse intraamygdaloid and intrahippocampal kainic acid (KA) models, and prevents epilepsy progression in the rat systemic KA model. We demonstrated that the robust antiepileptogenic effects of adenosine are based on an epigenetic mechanism, whereby increased levels of adenosine reduce global DNA methylation levels through mass action. It is now well-established that maladaptive changes in DNA methylation contribute to epileptogenesis and disease progression. Consequently, interrupting epigenetic processes that promote a hypermethylated state of DNA through therapeutic adenosine augmentation is antiepileptogenic. A key driving force for pathological increases in DNA methylation status are maladaptive changes in adenosine metabolism triggered by pathological overexpression of the key adenosine metabolizing enzyme ADK.
Related research articles by Dr. Detlev Boison:
- Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis
- Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice

Brain Regeneration after Traumatic Brain Injury and Stroke
To date, there is no therapy that can regenerate lost brain cells after a traumatic brain injury (TBI) or stroke. Whereas the immediate hours following a TBI or stroke might be crucial for long-term outcomes, neuroprotective strategies targeting this narrow time window have limited practical applicability. Attempts to help patients that suffered a TBI are therefore largely limited to rehabilitation strategies. Amazingly, the human brain is capable to gradually restore some of the lost functionality, indicating the existence of endogenous regenerative mechanisms. Those regenerative mechanisms are generally inhibited by inflammatory processes, but stimulated by a variety of neurotrophic factors including adenosine. In addition, stem cells that reside in the adult brain are capable to divide, if stimulated, and thereby to generate new nerve cells with new connections. Crucial for those endogenous regenerative capacities of the brain is the metabolic environment within the brain, which is largely defined by glial support cells, in particular astrocytes, which outnumber neurons 10:1 in certain areas of the human brain. Astrocytes regulate the availability of adenosine, which is an endogenous neuroprotectant and anti-inflammatory molecule that plays an important role in the neuro-regenerative capacities of the brain. Astrocytes are compromised after a brain injury, leading to deficiencies in adenosine metabolism and reduced capacity of the brain for repair. Therefore, therapeutic adenosine augmentation provides a neuroprotective environment and promotes endogenous repair processes.
Related research articles by Dr. Detlev Boison:
- Purines: Forgotten Mediators in Traumatic Brain Injury
- Caffeine prevents acute mortality after TBI in rats without increased morbidity
- Adenosine kinase determines the degree of brain injury after ischemic stroke in mice.
Comorbidities in Neurology
Comorbidities in Neurology represent a major conceptual and therapeutic challenge. For example, temporal lobe epilepsy (TLE) is a syndrome comprised of epileptic seizures and comorbid symptoms including memory and psychiatric impairment, depression, and sleep dysfunction. Similarly, Alzheimer’s disease (AD), Parkinson’s disease (PD), and Amyotrophic Lateral Sclerosis (ALS) are accompanied by various degrees of memory dysfunction. Patients with AD have an increased likelihood of seizures, whereas all four conditions share certain aspects of psychosis, depression, and sleep dysfunction. This remarkable overlap suggests common pathophysiological mechanisms, which include synaptic dysfunction and synaptotoxicity, as well as glial activation and astrogliosis. Astrogliosis is linked to synapse function via the tripartite synapse, but astrocytes also control the availability of gliotransmitters and adenosine.
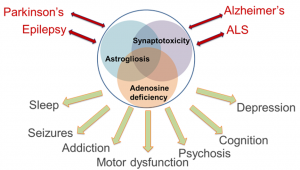
The ‘adenosine hypothesis of comorbidities’ implies that astrocyte activation, via overexpression of adenosine kinase (ADK), induces a deficiency in adenosine. Evidence from patient-derived samples shows astrogliosis and overexpression of ADK as a common pathological hallmark of epilepsy, AD, PD, and ALS. We have created and characterized a transgenic ‘comorbidity model’, in which brain-wide overexpression of ADK and resulting adenosine deficiency produces a comorbid spectrum of seizures, altered dopaminergic function, attentional impairment, and deficits in cognitive domains and sleep regulation. We conclude that dysfunction of adenosine signaling is common in neurological conditions, that adenosine dysfunction can explain co-morbid phenotypes, and that therapeutic adenosine augmentation might be effective for the treatment of comorbid symptoms in multiple neurological conditions.
To read Dr. Detlev Boison’s research, click here.
Metabolic Therapies
A ketogenic diet (KD) is an alternative metabolic treatment for epilepsy, and multiple retrospective and prospective studies confirm its clinical benefits. The KD’s high-fat low-carbohydrate composition forces ketone-based rather than glucose-based metabolism, but it is not known how this shift leads to anticonvulsant consequences. Primary applications of KD therapy include pediatric and medically refractory epilepsy; its use is increasing globally, and clinical benefits are similar across cultures and age groups. Despite its success, side effects and requisite strict compliance have limited widespread use of KDs, and a diet-based approach is often considered as a last resort. By better understanding the mechanisms involved in the anticonvulsant actions of a KD, pharmacological strategies might be developed that take advantage of beneficial aspects and limit problems associated with diet therapy. Adenosine acting at adenosine A1 receptors (A1Rs) is a logical candidate for the effects of KD therapy. Adenosine is well-established as a powerful anticonvulsant, and endogenous adenosine acting at A1Rs is an important seizure-control mechanism; deletion of A1Rs or increased adenosine clearance by elevated adenosine kinase (ADK) both cause spontaneous intrahippocampal electrographic seizures and increase the brain’s susceptibility to injury. Conversely, therapeutic adenosine augmentation is highly effective in controlling seizures. Adenosine is the core of ATP, a key molecule in basic biochemistry, and a ligand at its own family of G protein-coupled cell-surface receptors. Thus, adenosine is a homeostatic bioenergetic network regulator involved in metabolism and ongoing neuronal activity, and well-positioned to translate metabolic changes into altered brain activity. Using three lines of transgenic mice that all exhibit electrographic seizures due to deficient adenosine signaling we presented the first direct evidence that adenosine acting at A1Rs contributes to the therapeutic effects of KDs. Because KD augments adenosine signaling in the brain and because adenosine not only suppresses seizures but also affects epileptogenesis, we hypothesized that a ketogenic diet might prevent epileptogenesis through similar mechanisms. We tested this hypothesis in two independent rodent models of epileptogenesis. Using a pentylenetetrazole kindling paradigm in mice, we first show that a KD, but not a conventional antiepileptic drug (valproic acid), suppressed kindling-epileptogenesis. Importantly, after treatment reversal, increased seizure thresholds were maintained in those animals kindled in the presence of a KD, but not in those kindled in the presence of valproic acid. Next, we tested whether a KD can halt disease progression in a clinically relevant model of progressive epilepsy. Epileptic rats that developed spontaneous recurrent seizures after a pilocarpine-induced status epilepticus were treated with a KD or control diet (CD). Whereas seizures progressed in severity and frequency in the CD-fed animals, KD-fed animals showed a prolonged reduction of seizures, which persisted after diet reversal. KD-treatment was associated with increased adenosine and decreased DNA methylation, the latter being maintained after diet discontinuation. Our findings demonstrate that a KD prevented disease progression in two mechanistically different models of epilepsy, and suggest an epigenetic mechanism underlying the therapeutic effects.
Related research articles by Dr. Detlev Boison:
- A ketogenic diet suppresses seizures in mice through adenosine A1 receptors
- Ketogenic Diet Prevents Epileptogenesis and Disease Progression in Adult Mice and Rats
- New insights into the mechanisms of the ketogenic diet


Detlev Boison, PhD
Professor & Vice Chair of Research & Training, Department of Neurosurgery