Research
In proliferating cells, biosynthetic requirements vary across cell cycle phases, indicating that phase-specific control of cellular metabolism is essential. This raises fundamental questions: How does cellular metabolism differ from one cell cycle phase to another? How does the cell cycle machinery mechanistically link to changes in metabolism? What is the significance of phase-specific metabolic regulation for cell growth and proliferation, human health and disease? Despite the critical importance of these questions, we currently have very few answers. To truly understand cell growth and proliferation, we must gain a deep understanding of the underlying metabolic changes that are orchestrated throughout the cell cycle. Such understanding could also present a wealth of new therapeutic opportunities in diseases such as cancer.
Although large-scale metabolic reprogramming is a ubiquitous hallmark of cancer cells, activating mutations or amplifications of metabolic enzymes themselves are rare in cancer. Instead, cancer cells typically stimulate anabolism by co-opting the signaling nodes that control large metabolic networks. This allows cancer cells to coordinately upregulate many metabolic pathways that must function cooperatively to produce complex biomass, rather than upregulating a single metabolic enzyme or pathway in isolation. Many of the most common oncogenic driver mutations uncouple these signaling and metabolic outputs from their normal regulatory inputs. Although this provides a growth advantage, locking pathways in the “on” state can come at the cost of reduced plasticity and increased dependence on specific nutrients, enzymes or pathways for sustained growth and viability. Thus the uniquely reprogrammed metabolic networks in cancer cells offer opportunities to identify and target metabolic processes that are uniquely essential in those cells.
Thus we are defining cell cycle phase-specific control of cellular metabolism by studying metabolic regulation in individual cell cycle phases, and using that information to identify and exploit new phase-specific metabolic vulnerabilities in cancer.
Current projects include:
Defining cell cycle phase-specific control of metabolism
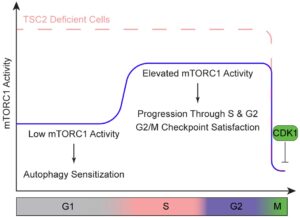 We recently discovered that the activity of the master metabolic regulator mTOR Complex 1 (mTORC1), oscillates throughout the cell cycle, with important functional consequences for cell growth and proliferation (Joshi et al., bioRxiv). This oscillation is mediated through multiple mechanisms, including differential regulation of an essential upstream negative regulator of mTORC1 called the TSC complex throughout interphase, and CDK1-dependent effects on mTORC1 itself during mitosis. Elevated mTORC1 activity in S and G2 is important for progression through those phases, and low mTORC1 activity in G1 sensitizes cells to induction of autophagy. We are now defining cell cycle phase-specific functions of mTORC1 in regulating metabolism, along with mTORC1-independent changes that are critical for cell cycle progression.
We recently discovered that the activity of the master metabolic regulator mTOR Complex 1 (mTORC1), oscillates throughout the cell cycle, with important functional consequences for cell growth and proliferation (Joshi et al., bioRxiv). This oscillation is mediated through multiple mechanisms, including differential regulation of an essential upstream negative regulator of mTORC1 called the TSC complex throughout interphase, and CDK1-dependent effects on mTORC1 itself during mitosis. Elevated mTORC1 activity in S and G2 is important for progression through those phases, and low mTORC1 activity in G1 sensitizes cells to induction of autophagy. We are now defining cell cycle phase-specific functions of mTORC1 in regulating metabolism, along with mTORC1-independent changes that are critical for cell cycle progression.
Targeting ribonucleotide (NTP) synthesis pathways to selectively kill cancer cells
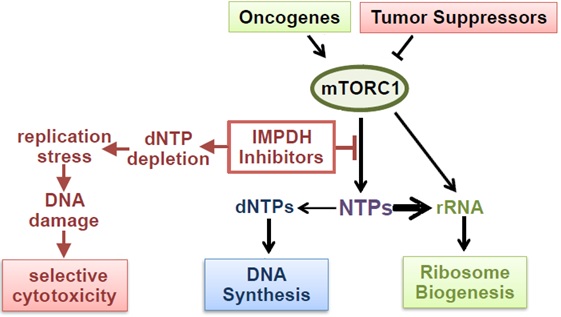
Maintaining intracellular nucleotide pools is essential for cell proliferation and viability. We discovered that tumor cells with high rates of ribosomal RNA (rRNA) synthesis are uniquely dependent on nucleotide synthesis pathways to maintain their free nucleotide pools and sustain rapid rRNA and DNA synthesis (Valvezan et al., 2017, Cancer Cell). This metabolic dependency can be exploited using clinically approved inhibitors of the rate-limiting enzyme in de novo guanylate nucleotide synthesis (IMPDH), which cause nucleotide depletion and apoptosis selectively in tumor cells with elevated rRNA synthesis rates. In cell and tumor models of the genetic tumor syndrome Tuberous Sclerosis Complex (TSC), IMPDH inhibitors demonstrate strong anti-tumor efficacy at clinically relevant doses (Valvezan et al., 2017, Cancer Cell; Valvezan et al., 2020, JCI Insight).
TSC tumors are driven by uncontrolled activation of the master metabolic regulator mTOR Complex 1 (mTORC1). We discovered that uncontrolled mTORC1 activation confers dependence on nucleotide synthesis pathways by strongly driving rRNA synthesis. As mTORC1 is activated in the majority of human tumors across nearly all lineages, we are expanding these nucleotide synthesis inhibitor studies to determine the full translational potential of this targetable metabolic vulnerability. IMPDH inhibitors have been used clinically for decades as safe and well-tolerated immunosuppressants, but are not currently used as anti-tumor agents. By contrast, direct inhibitors of dNTP synthesis and DNA synthesis have been used as chemotherapeutic agents for decades, but are associated with significant toxicity which can limit their efficacy. By targeting NTP synthesis rather than dNTP/DNA synthesis, IMPDH inhibitors have increased specificity for targeting tumor cells with high rates of rRNA synthesis, which corresponds to their markedly reduced toxicity profiles in patients. The use of clinically approved IMPDH inhibitors in our studies means that efficacy could lead to rapid repurposing for cancer patients.
Defining and targeting metabolic reprogramming in colorectal cancer
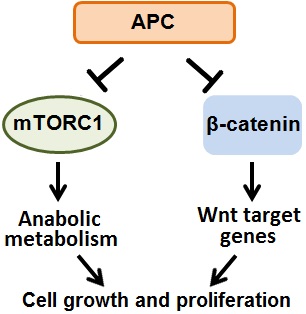
Colorectal cancer is the 2nd leading cause of cancer-related death in the United States, and is responsible for over 800,000 deaths globally each year. Sporadic truncating mutations in the APC tumor suppressor gene promote the onset of 80% of human colorectal cancers, and germline APC mutations cause the intestinal polyposis and colorectal cancer predisposition syndrome Familial Adenomatous Polyposis, with 100% penetrance. Thus it is imperative to understand the role of APC in cellular signaling and metabolism, and the consequences of APC mutations in colorectal cancer. However, despite their potent tumorigenic effects, very little is known about how APC mutations alter cellular metabolism. APC mutations activate the canonical Wnt/β-catenin signaling pathway and in parallel also activate the master metabolic regulator mTOR Complex 1. We are defining the metabolic consequences of APC mutations in colorectal cancer with the goal of identifying and exploiting unique metabolic dependences. Intestinal nutrients and the microbiota provide a particularly interesting and complex microenvironment compared to many other solid tumors which are exclusively fed by serum metabolites. Thus we are also determining how the cell intrinsic effects of APC mutations and the intestinal tumor microenvironment interact to produce the complex metabolic phenotype of colorectal cancer cells.