Findings
Genome sequencing and analysis of the rice coral Montipora capitata
Alexander Shumaker, Hollie Putnam, Huan Qiu, Udi Zelzion, Dana C. Price, Nicole Wagner, Arik Harel, Ruth Gates, Hwan Su Yoon, and Debashish Bhattacharya
Ongoing efforts to conserve coral reefs by identifying the major stress response pathways and thereby laying the foundation to select resistant genotypes rely on a robust genomic foundation. We recently generated a high quality long-read based ~886 million base pair (Mbp) nuclear genome assembly and transcriptome data from the dominant rice coral, Montipora capitata from Hawai’i. Our work provides insights into the architecture of coral genomes and shows how they differ in size and gene inventory, we postulate that significant population size reduction explains the large genome size in M. capitata. The Hawaiian population represents an endemic with reduced standing genetic variation that has resulted in the expansion of parasitic genetic elements such as transposons. We also describe a recent example of foreign gene acquisition via a bacterial gene transfer agent (GTA; see figure). GTAs are small segments of DNA (ca. 4 kbp in size) that move between cells in a bacterial population, but here we describe a GTA that has moved between kingdoms. This result identifies a pathway for bacterial gene integration into coral nuclear genomes, something we have described previously in a taxonomically broad analysis of coral genomic data (Bhattacharya et al. 2016; eLife DOI:10.7554/eLife.13288). Our work also illustrates the major pathways of stress response that can be used to predict regulatory components of the transcriptional networks in M. capitata. These genomic resources provide insights into the adaptive potential of sessile, long-lived coral species in both natural and human influenced environments. Ultimately, our results will be used to support functional and population genomic studies aimed at Hawaiian reef restoration and conservation.
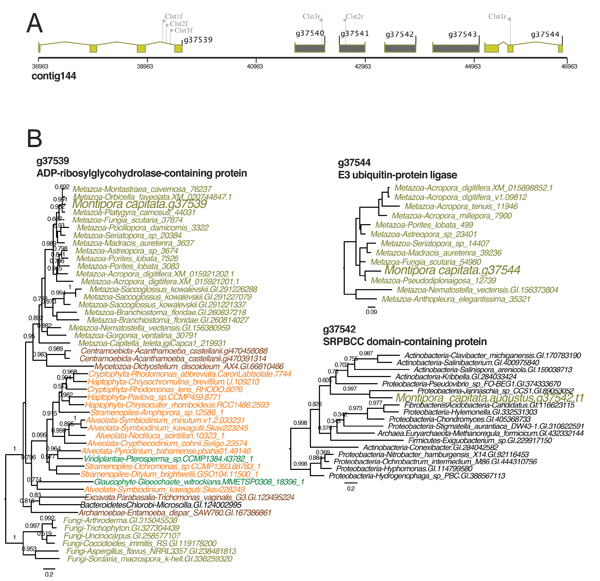
GTA integration in the M. capitata genome
A) Location of the bacterium-derived 4-gene GTA cluster in M. capitata nuclear genome contig144. The coral (animal) genes and GTA candidates are shown in light brown and dark grey colored filled boxes, respectively.
B) Phylogenetic trees for one of the four GTA-derived genes (g37542) and the flanking coral genes validating the bacterial origin of the GTA region. Branch support (larger than 0.5) are local support values estimated using the Shimodaira-Hasegawa test.
Spread of complex structured repeats in stony corals
Huan Qiu, Ehud Zelzion, Hollie M. Putnam, Ruth D. Gates, Nicole E. Wagner, Diane K. Adams, Debashish Bhattacharya
How stony corals (Scleractinia), a 240 million-year-old lineage, have survived at least five major mass extinctions, despite their apparent vulnerability to environmental insults (e.g., mass bleaching) is a puzzling and unexplained observation. Previous work has focused on population genetics, biogeography, and analysis of protein coding regions to find clues to the basis of coral resilience. Here we took a novel approach by analyzing non-coding DNA and made a surprising finding. We report a novel class of repeated DNAs in stony corals that may play a role in adaptation by these key ecosystem engineers. Our analysis of assembled RNA-seq data from the robust coral Montipora capitata turned up conserved repeat families (named SCORs) in the untranslated regions (UTRs), all of which form long, stable RNA hairpins (see figure). SCORs do not encode known micro-RNAs or long non-coding RNAs in corals. Analysis of the complete genome of the complex coral Acropora digitifera and genomic data from 18 other coral species revealed that many SCORs are highly conserved at the sequence level and widely distributed in different frequencies in the genome. Genome-wide analysis shows that SCORs are also present in introns, although the majority is located in intergenic regions. SCORs have undergone frequent and rapid duplication and degradation, and horizontal transfer within and between coral species. We postulate that SCOR movement is likely dependent on reverse transcription and integration, aided by the abundant retroelements present in coral genomes. Until now, we find no evidence that gene-associated SCOR expression correlates with pathways associated with development, symbiosome formation, indicating that they do not regulate these highly conserved, core coral functions, but may play a regulatory role in other pathways. We speculate that due to their surprisingly high sequence identities across deeply diverged corals, physical association with genes, and dynamic evolution, SCORs may have adaptive functions in corals that need to be explored using population genomic and function-based approaches.
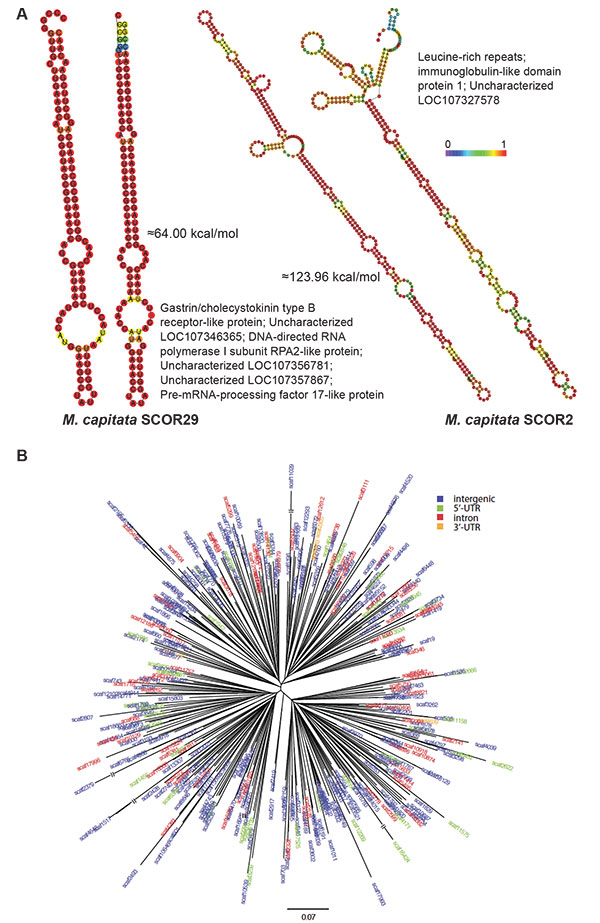
Analysis of coral SCORs.
A) Predicted minimum free energy (MFE) secondary structures of SCOR29 and SCOR2 from two different genomic locations in M. capitata are shown, as well as a list of other genes with which they are associated in this coral. The structures are colored according to base-pairing probabilities, with red being more stable. For regions not involved in pairing, the color denotes the probability of being unpaired. The MFEs for two structures are indicated.
B) Phylogenetic tree of an Acropora digitifera SCOR family (Adig.CORrep 17). Repeats located in different genomic regions are shown in different colors.