Phylogenetic Tree Construction
by Celeste Zhang
Phylogenetic trees show the proposed evolutionary relationships between various organisms. When isolating a new strain of microbe, it is usually recommended to create a phylogenetic tree to assess how the strain relates to other species of the same genus/family or even other strains of the same species. This is especially important when strains of the same species or species of the same genus are very closely related and cannot be easily discerned with an NCBI BLAST search (e.g. members of the Pseudomonas genus).
This page is meant to be a tutorial for creating your own phylogenetic tree using the MEGA11 software. For more information on the principles and explanations behind each step, please visit: Building Phylogenetic Trees from Molecular Data with MEGA by Barry G. Hall
This tutorial assumes you have already sequenced your gene of interest from your microbe of interest; this gene will be referred to as your “query sequence.”
1. Obtaining homologous DNA sequences:
In order to have a phylogenetic tree, you need to have homologous sequences to compare your query sequence to. In order to obtain those sequences, you will need to do an NCBI BLAST search to find similar sequences that have been entered into GenBank.
- Go to the NCBI BLAST website
- Click nucleotide BLAST for comparison of nucleotide sequences (page may change over time, so the UI may be different)
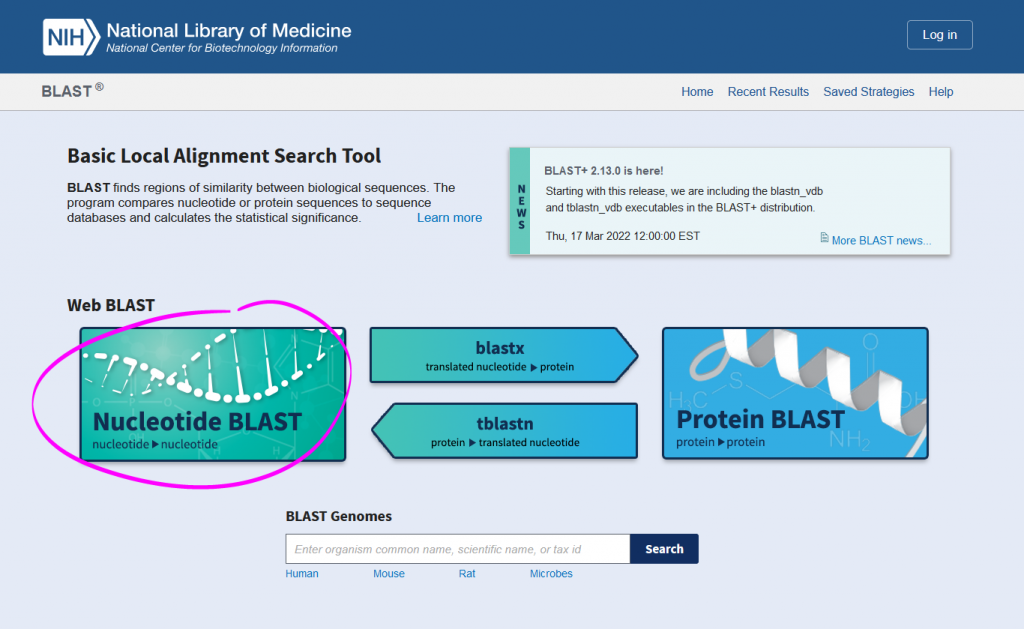
- Enter your query sequence in the textbox under “Enter accession number(s), gi(s), or FASTA sequence(s)”
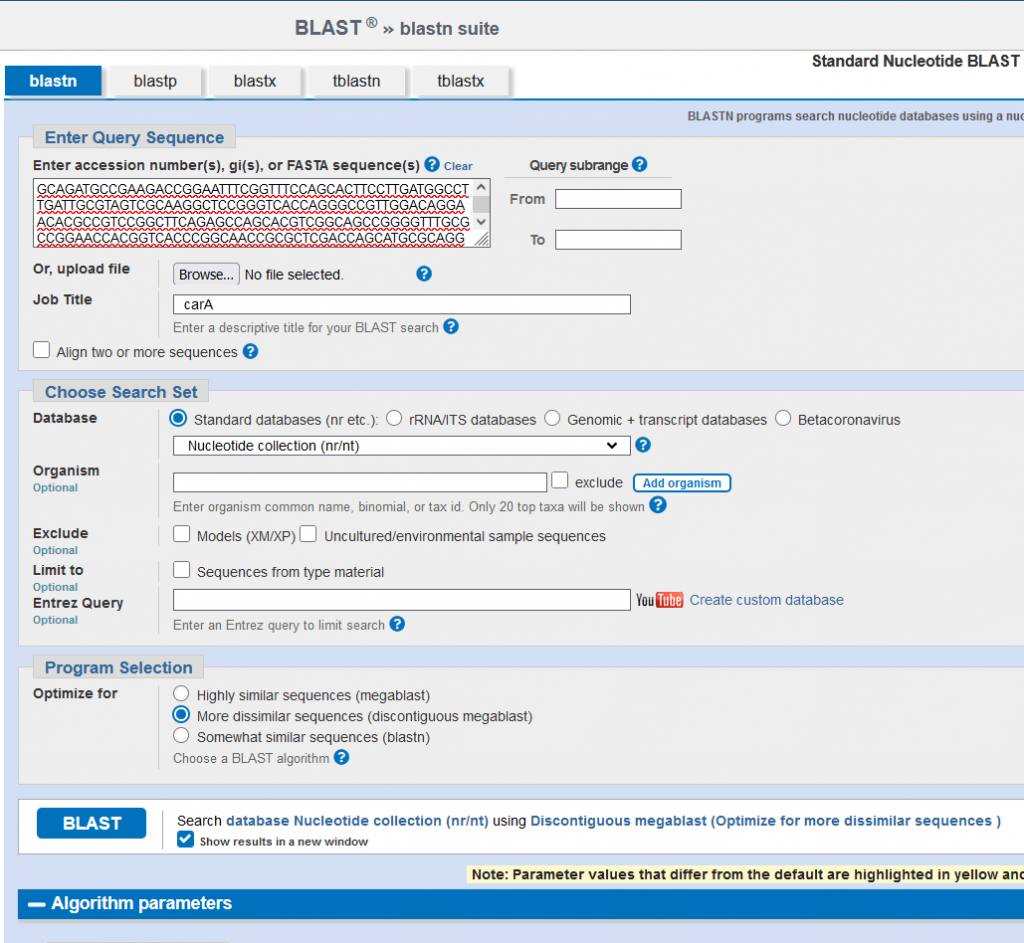
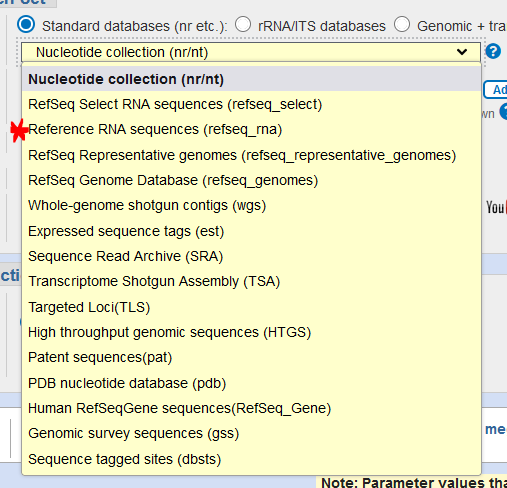
- Choose your database
- For most genes, the Standard databases – Nucleotide collection is optimal
- However, for 16S rRNA/ITS sequences, you can pick the “rRNA/ITS database”, or the “Standard databases – Reference RNA sequences”. These databases hold representative sequences for the 16S rRNA/ITS gene(s) of a given species
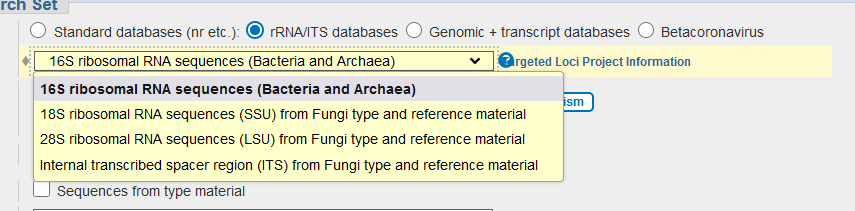
- Select your BLAST program
- “Highly similar sequences” will give you the sequences that are most closely related to your query sequences (good for quick identification of a strain, usually only really helpful for a genus-level ID since there will be many matches above 95% similarity)
- “More dissimilar sequences” will give you sequences that are not as alike to the query sequence (good for making a tree that shows a variety of species and how your target strain relates to those species, since there will be fewer high-similarity matches)
- OPTIONAL for genera/species that generate many matches above 95% similarity (ex. Pseudomonas): click “Algorithm Parameters” and change “Max target sequences”
- Depending on how many closely related matches your genus/species tends to get, change the number from 100 to 500-1000
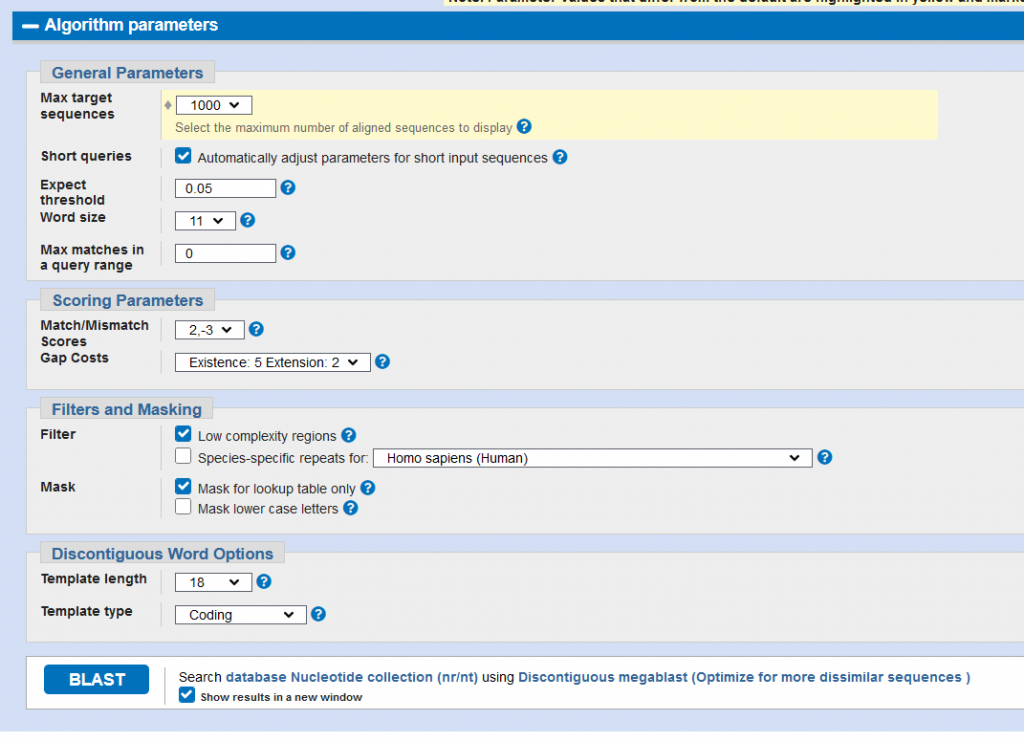
- Hit BLAST to get your results
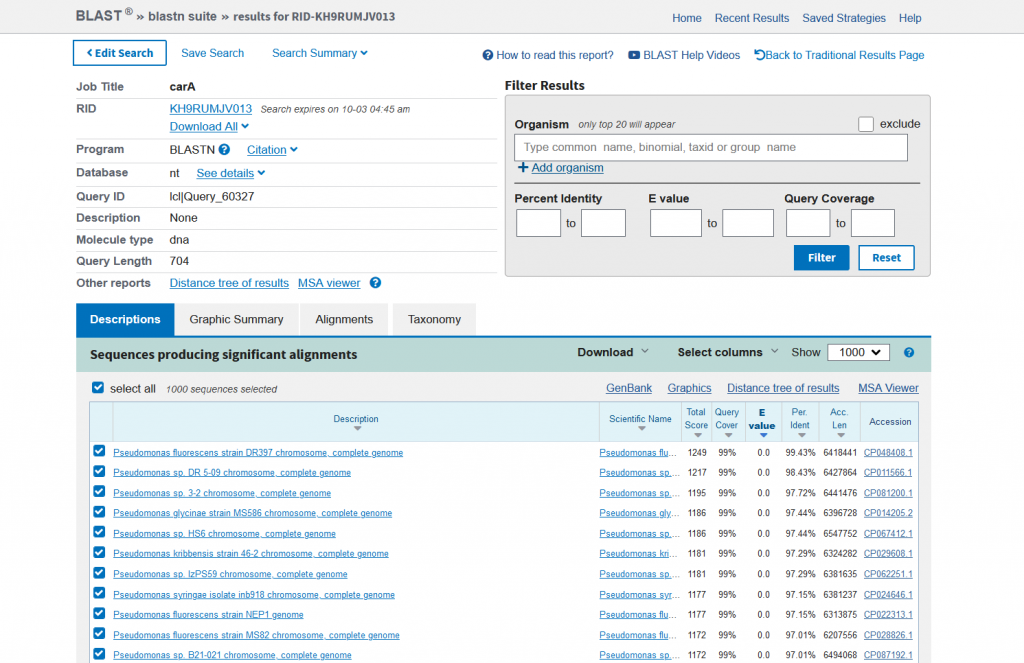
- You will need to select about 20-30 strains to create your phylogenetic tree with
- Unselect all sequences and manually pick sequences – this part is the most important!
- Don’t just pick the most identical sequences (90-100% identity), you’ll also need some outgroups at <90% identity
- Make sure you have a good spread of sequences to have the best tree possible
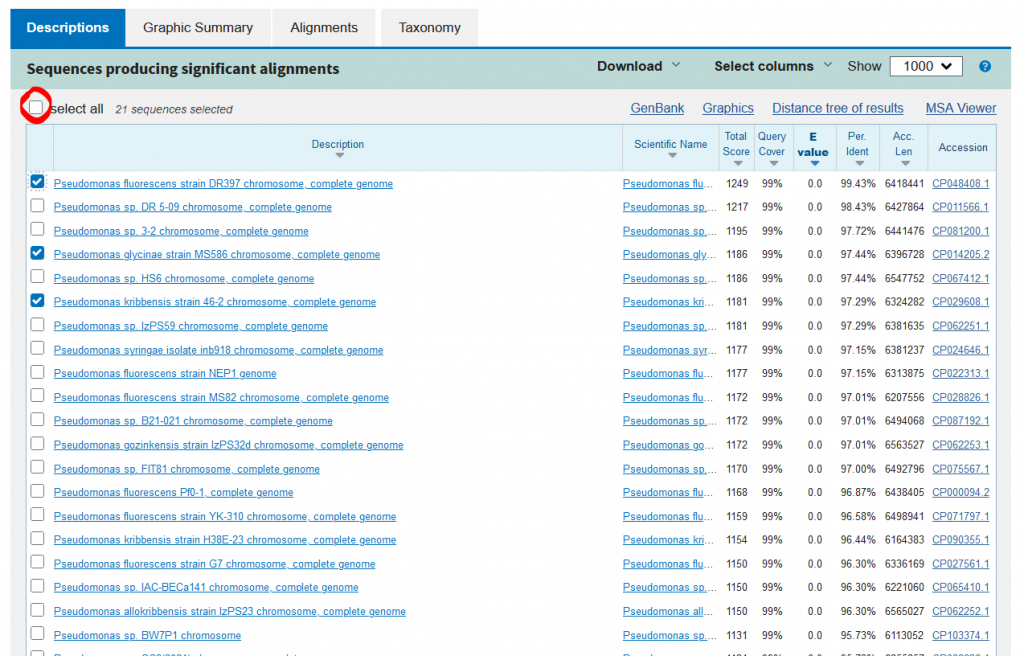
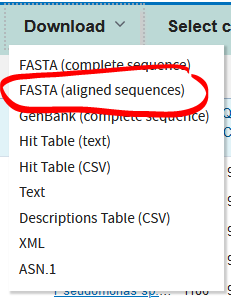
- Once you’re done selecting sequences, go to download and click FASTA (aligned sequences), which will download a .txt file
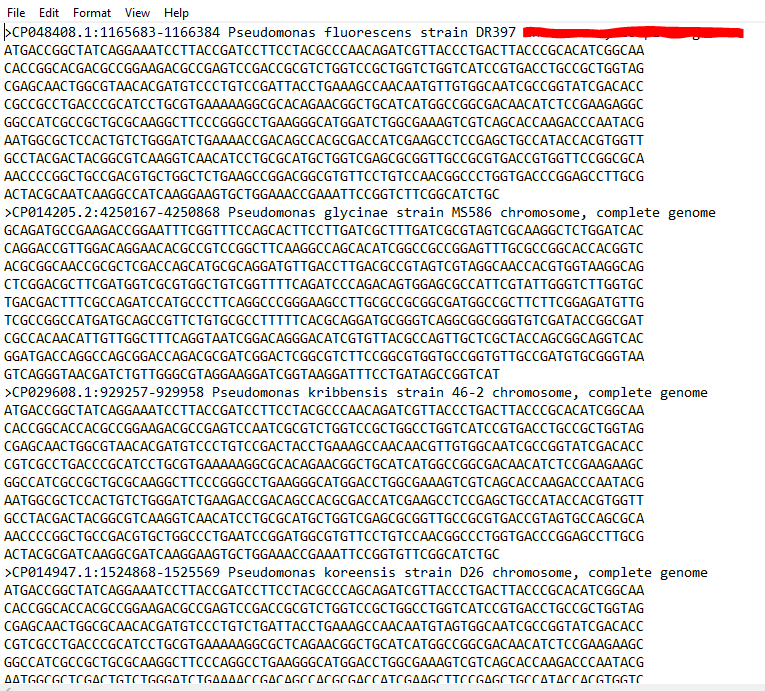
- Open the .txt file, which will show the sequences in FASTA format
- Add your sequences in FASTA format:
>description (usually “Genus species strain x”)
SEQUENCE

- Make any changes, such as deleting “chromosome, complete genome” or other repetitive additions to the names, but leave behind the strain names
- Save the file as a .fasta file so that MEGA11 will be able to understand it
2. Aligning your sequences:
- Open up MEGA11 and go to “ALIGN – Edit/Build Alignment”
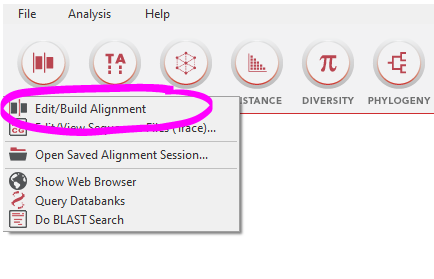
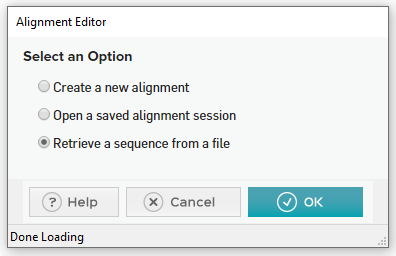
- Select “Retrieve a sequence from a file” and select the .fasta you made earlier
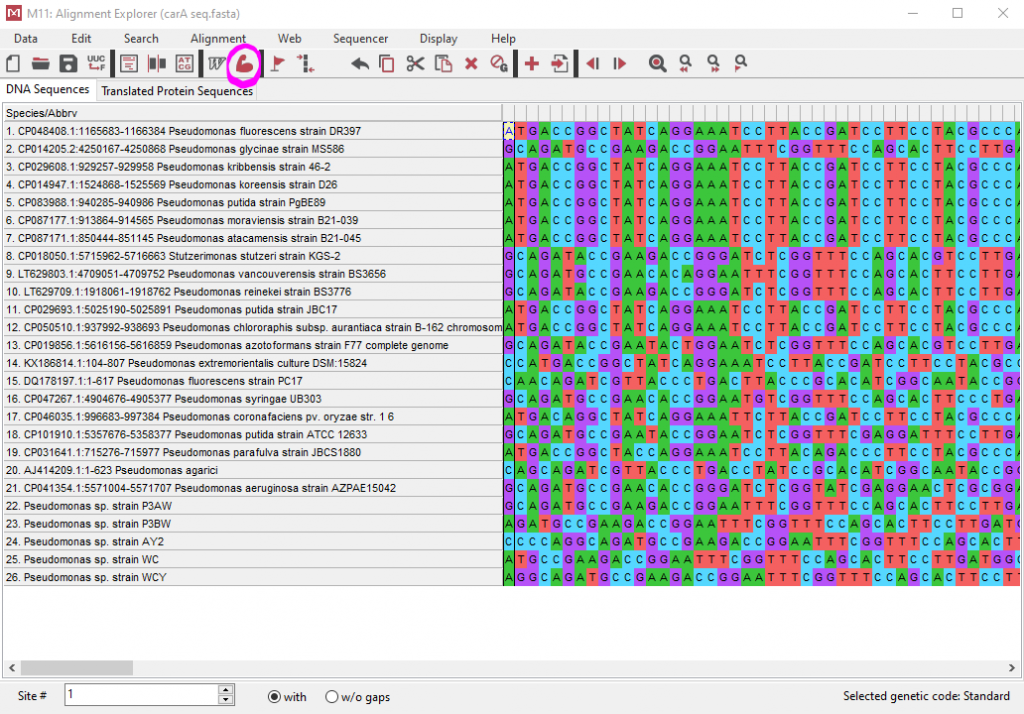
- Click “Alignment – Align by MUSCLE” or hit the red 💪 emoji
- Don’t mess with the default options and just click OK
- Once everything is aligned, go to “Data – Export Alignment – MEGA Format” and save as a MEG file
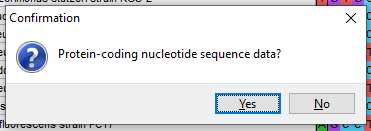
- The program will ask you if it’s protein-coding – for 16S rRNA, the answer is No, but for other genes it may be Yes
3. Generating the tree:
- For this tutorial, we will be making a Maximum Likelihood (ML) tree
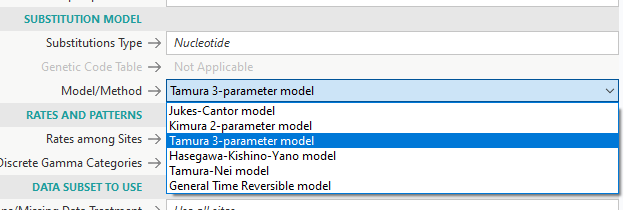
- Click on “MODELS – Find Best DNA/Protein Models (ML)” so that the program can figure out which type of substitution model is best for your data
- Don’t change any default settings and click OK
- The program will generate a chart showing which how each model fits (lower BIC is better)
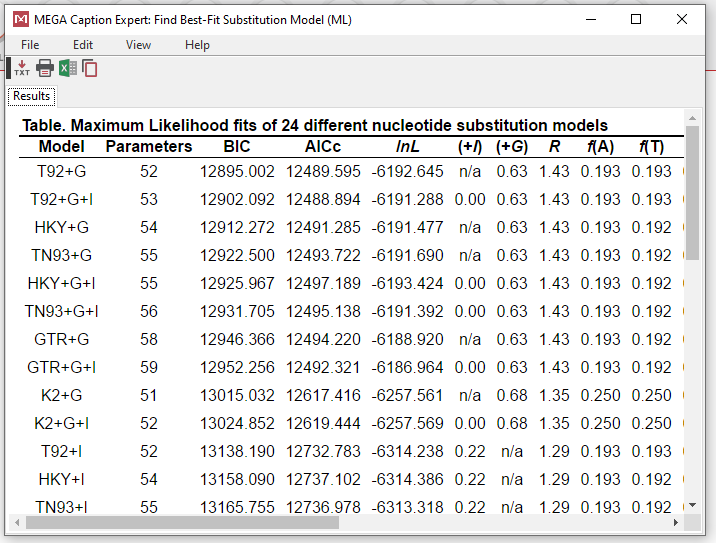
- K2 = Kimura 2-parameter
- T92 = Tamura 3-parameter
- TN93 = Tamura-Nei
- HKY = Hasegawa-Kishino-Yano
- GTR = General Time Reversible
- JC = Jukes-Cantor
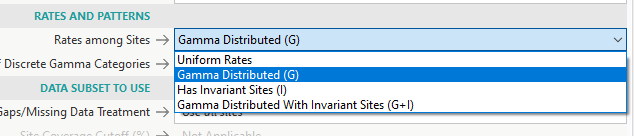
- +G = gamma distributed
- +I = has invariant sites
- +G+I = gamma distributed with invariant sites
- Go to “PHYLOGENY – Construct/Test Maximum Likelihood Tree”
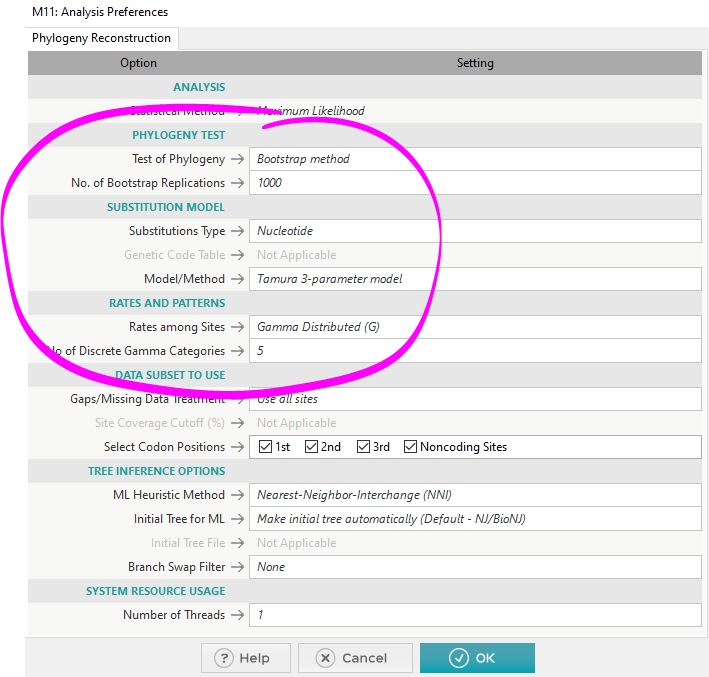
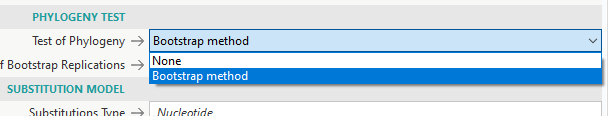
- For Phylogeny Test, there are 2 options: None or Bootstrap
- None is for quick generation of 1 tree
- Bootstrap involves simulations of several trees to test the stability/accuracy of each branch of the final tree
- If you want to publish the tree in a paper, you need to pick Bootstrap with at least 1000 replications (500 might be okay)
- Substitution Model is where you have to pick the model that best adheres to your data from the model you generated earlier (T92/K2/TN93/etc.)
- Rates and Patterns is where you have to pick +G/+I/+G+I, if your model indicated that you should pick those options
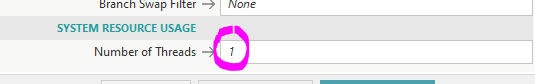
- For MEGA11, using more than a single thread (System Resource Usage) will sometimes crash the program!
- Using a single thread is safe, but slow
- Using more than 1 will be faster, but risks crashing partway into the simulation
- Click OK and wait
- Doing 500-1000 reps takes a while, for instance it takes my desktop with an Intel DDR4 i512600K CPU ~45 min to generate 1000 reps with a T92+G model
HKY, TN93, and GTR are the slowest
- Doing 500-1000 reps takes a while, for instance it takes my desktop with an Intel DDR4 i512600K CPU ~45 min to generate 1000 reps with a T92+G model
4. Editing the tree display:
- Once the tree has been generated, this window will pop up
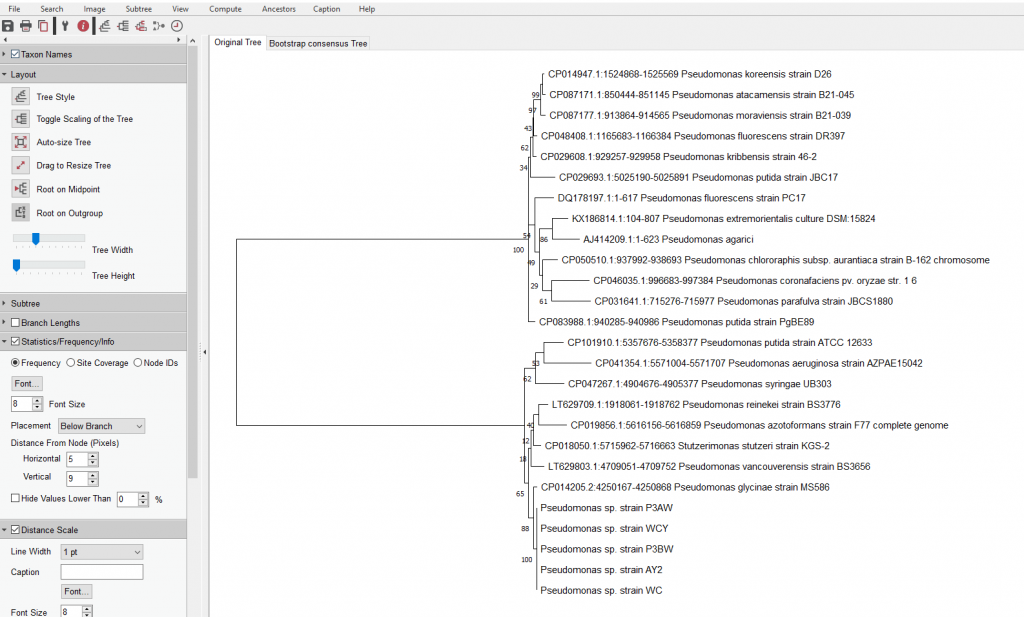
- As you can see, this tree is quite hard to understand since all the branches are so close together
- Go to “Layout” and play with the “Tree Width” option to scale the width of the tree
- If you did the bootstrap method, you’ll see these numbers at the branches
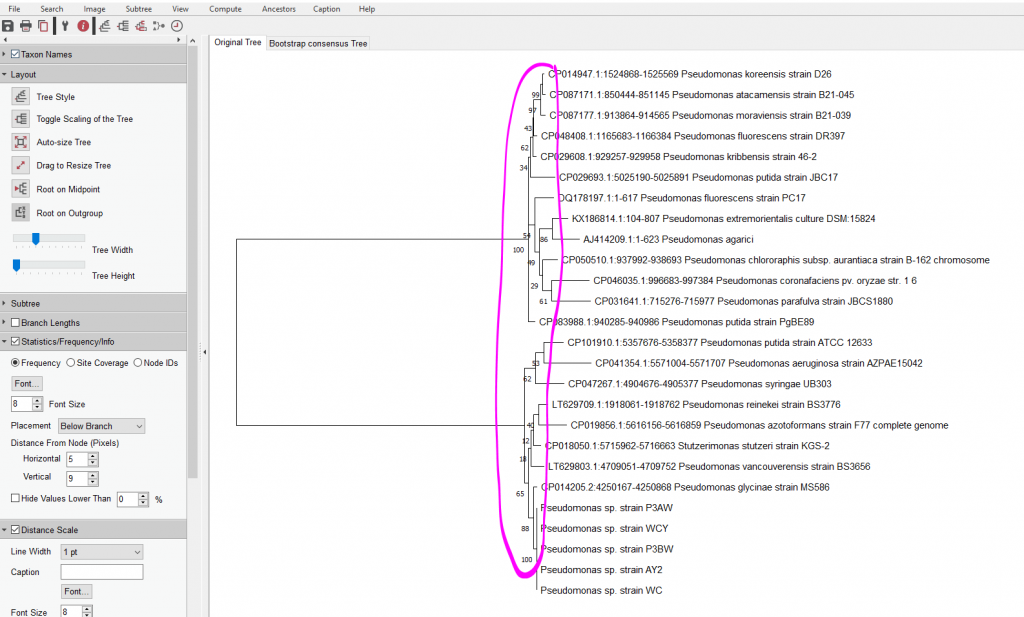
- These numbers represent the percentage of times these branches appeared in those positions over the course of your 1000 reps
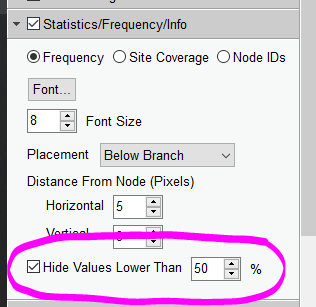
- Anything under 70 is considered suspect, but many papers will only hide numbers under 50
- To hide values lower than 50, go to “Statistics/Frequency/Info”, check the option “Hide Values Lower Than __%”, and input 50
- Once you’re happy with your tree, go to Image and select your format of choice
- You might also want to save the tree in a format MEGA11 can understand, just in case you have to go back to it and don’t want to waste more time re-generating the tree